Abstract
Background: Real-time PCR analysis is a sensitive DNA quantification technique that has recently gained considerable attention in biotechnology, microbiology and molecular diagnostics. Although, the cycle-threshold (Ct) method is the present "gold standard", it is far from being a standard assay. Uniform reaction efficiency among samples is the most important assumption of this method. Nevertheless, some authors have reported that it may not be correct and a slight PCR efficiency decrease of about 4% could result in an error of up to 400% using the Ct method. This reaction efficiency decrease may be caused by inhibiting agents used during nucleic acid extraction or copurified from the biological sample. We propose a new method (Cy0) that does not require the assumption of equal reaction efficiency between unknowns and standard curve.Results: The Cy0 method is based on the fit of Richards' equation to real-time PCR data by nonlinear regression in order to obtain the best fit estimators of reaction parameters. Subsequently, these parameters were used to calculate the Cy0 value that minimizes the dependence of its value on PCR kinetic.
The Ct, second derivative (Cp), sigmoidal curve fitting method (SCF) and Cy0 methods were compared using two criteria: precision and accuracy. Our results demonstrated that, in optimal amplification conditions, these four methods are equally precise and accurate. However, when PCR efficiency was slightly decreased, diluting amplification mix quantity or adding a biological inhibitor such as IgG, the SCF, Ct and Cp methods were markedly impaired while the Cy0 method gave significantly more accurate and precise results.
Conclusion: Our results demonstrate that Cy0 represents a significant improvement over the standard methods for obtaining a reliable and precise nucleic acid quantification even in sub-optimal amplification conditions overcoming the underestimation caused by the presence of some PCR inhibitors.






Hello, You are the visitor, Thanks.
![]()
Visiting us from ...
Warning: mysqli_real_escape_string() expects parameter 1 to be mysqli, boolean given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 43
Notice: Only variables should be passed by reference in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 44
Warning: mysqli_query() expects parameter 1 to be mysqli, boolean given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 65
Warning: mysqli_num_rows() expects parameter 1 to be mysqli_result, null given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 66
Warning: mysqli_fetch_assoc() expects parameter 1 to be mysqli_result, null given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 68
Warning: mysqli_query() expects parameter 1 to be mysqli, boolean given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 85
Warning: mysqli_num_rows() expects parameter 1 to be mysqli_result, null given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 86
Warning: file_get_contents(http://www.geoplugin.net/json.gp?ip=216.73.216.152): failed to open stream: HTTP request failed! HTTP/1.1 403 Forbidden in /home/cymethod/domains/cy0method.org/public_html/functions/geoIP.db-ip.com.php on line 37
Warning: get_object_vars() expects parameter 1 to be object, null given in /home/cymethod/domains/cy0method.org/public_html/functions/geoIP.db-ip.com.php on line 37
latest clicks ...
Warning: mysqli_query() expects parameter 1 to be mysqli, boolean given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 150
Warning: mysqli_num_rows() expects parameter 1 to be mysqli_result, null given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 157
Warning: mysqli_fetch_array() expects parameter 1 to be mysqli_result, null given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 163
Warning: mysqli_close() expects parameter 1 to be mysqli, boolean given in /home/cymethod/domains/cy0method.org/public_html/navigation/last_5_clicks.php on line 174
Background
In the last few years, the real-time polymerase chain reaction (PCR) has rapidly become the most widely used technique in modern molecular biology [1-4].This technique relies on fluorescence-based detection of amplicon DNA and allows the kinetics of PCR amplification to be monitored in real time, making it possible to quantify nucleic acids with extraordinary ease and precision. With a large dynamic range (7-8 magnitudes) and a high degree of sensitivity (5-10 molecules), the real-time PCR addresses the evident requirement for quantitative data analysis in molecular medicine, biotechnology, microbiology and diagnostics [ 5, 6].
Methods
Experimental design
The absolute quantification method relies on the comparison of distinct samples, such as the comparison of a biological sample with a standard curve of known initial concentration [21].We wondered how accuracy and precision change when a standard curve is compared with unknown samples characterized by different efficiencies. A natural way of studying the effect of efficiency differences among samples on quantification would be to compare the amounts of a quantified gene.
A slight amplification inhibition in the quantitative real-time PCR experiments was obtained by using two systems: decreasing the amplification mix used in the reaction and adding varying amounts of IgG, a known PCR inhibitor.
For the first system, we amplified the MT-ND1 gene by real-time PCR in reactions having the same initial amount of DNA but different amounts of SYBR Green I Master mix. A standard curve was performed over a wide range of input DNA (3.14x107-3.14x101) in the presence of optimal amplification conditions (100% amplification mix), while the unknowns were run in the presence of the same starting DNA amounts but with amplification mix quantities ranging from 60% to 100%. This produced different reaction kinetics, mimicking the amplification inhibition that often occurs in biological samples [17, 22].
Furthermore, quantitative real-time PCR quantifications were performed in the presence of an optimal amplification reaction mix added with serial dilutions of IgG (0.0625 - 2 µg/ml) thus acting as the inhibitory agent [23].
The reaction efficiency obtained was estimated by the LinReg method [24]. This approach identifies the exponential phase of the reaction by plotting the fluorescence on a log scale. A linear regression is then performed leading to the estimation of the efficiency of each PCR reaction.
Quantitative Real-Time PCR
The DNA standard consisted of a pGEM-T (Promega) plasmid containing a 104 bp fragment of the mitochondrial gene NADH dehydrogenase 1 (MT-ND1) as insert. This DNA fragment was produced by the ND1/ND2 primer pair (forward ND1: 5 ' -ACGCCATAAAACTCTTCACCAAAG-3 ' and reverse ND2: 5 ' -TAGTAGAAGAGCGATGGTGAGAGCTA-3 ' ). This plasmid was purified using the Plasmid Midi Kit (Qiagen) according to the manufacturer ' s instructions. The final concentration of the standard plasmid was estimated spectophotometrically by averaging three replicate A260 absorbance determinations.Description of the SCF method
Fluorescence readings were used to fit the following 4-parameter sigmoid function using nonlinear regression analysis:|
|
Eq. 1 |
|
|
Eq. 2 |
Description of the Cy0 method
The Cy0 value is the intersection point between the abscissa axis and tangent of the inflection point of the Richards curve obtained by the non-linear regression of raw data (Fig. 1).
Figure 1
Example of modelling PCR amplification with a 5-parameter Richards function Effectiveness of this model is illustrated by the predicted values generated by Eq. 3 (open circles) that agree with the observed fluorescence (dot and line). Curve-fitting of experimentally derived fluorescence dataset to Eq. 3 generates values for the kinetic parameters from which the inflection point (solid black rhombus) and the slope of the curve can be derived. The quantitative entity Cy0 (solid black dot), used in the proposed method, shows the cross point between the x-axis and the tangent crossing the inflection point of real-time PCR fluorescence curve.|
|
Eq. 3 |
|
|
Eq. 4 |
|
|
Eq. 5 |
|
|
Eq. 6 |
Statistical data analysis
Nonlinear regressions (for 4-parameter sigmoid and 5-parameter Richards functions) were performed determining unweighted least squares estimates of parameters using the Levenberg-Marquardt method. Accuracy was calculated using the following equation:Results
Experimental system 1: reduction of amplification mix percentage
With our experimental set up, the mean PCR reaction efficiency was 88% under optimal amplification conditions and slightly decreased in the presence of smaller amplification mix up to 84%. Moreover, for decreasing amplification mix amounts, the PCR reaction efficiencies showed higher dispersion levels than optimal conditions leading to increasing quantitative errors (Variation Interval, VI100%= 1.921-1.852 and VI60%= 1.903-1.776; Fig. 2). Subsequently, the fluorescence data obtained in these reactions were used to calculate the initial DNA amount using four different procedures: SCF, Ct, Cp and Cy0.Precision and accuracy of the SCF method
Previous studies have shown that the SCF approach can lead to quantification without prior knowledge of amplification efficiency [18, 19, 26]; therefore, we evaluated the performance of this method on our data set. To assess the effect of unequal efficiencies on accuracy, the calculated input DNA, expressed as molecular number, was compared to the expected value obtaining the relative error (RE). The precision was further evaluated measuring the variation coefficient (CV%) of the estimated initial DNA in the presence of different PCR efficiencies and input DNA.In our experimental design, the SCF method showed a very poor precision (mean CV% = 594.74%) and low accuracy (mean RE = -5.05). The impact of amplification efficiency decline on accuracy was very strong resulting in an underestimate of samples of up to 500% (Additional file 3). The log transformation of fluorescence data before sigmoidal fitting significantly reduced the CV% and RE to 66.12% and -0.20, respectively; however, the overall bias remained the same [19]. Finally, we also tested an improved SCF approach based on a previous study by Rutledge 2004 [26] without obtaining significant amelioration (Additional file 4).

Figure 2
Estimation of PCR efficiency using LinReg method Efficiency values were determined from 420 independent reactions using a combination of 3.14 x 107 x 3.14 x 101 DNA molecules as starting template and amplification mix quantities ranging from 60% to 100%. The graph shows the distribution of PCR efficiencies in relation to the percentage of amplification mix used in the reaction. The solid black squares (?) represent the mean of each distribution.The Cy0 method
The SCF model assumes that the fluorescence signal is proportional to the amount of product, which is often the case for SYBR-Green I real-time PCR performed with saturing concentrations of dye. In such conditions, centrally symmetric amplification curves are expected. However, in our experience, we found several non-symmetric amplification curves shown to have good amplification efficiency using standard curve analysis (Additional file 1 and 3). In order to find a suitable mathematical representation of the complete PCR kinetic curve we compared the standard error of estimate obtained by several equations that generate S-shaped curves (Tab. 1). As shown in Figure 1, these results demonstrated that real-time PCR readouts can be effectively modelled using the 5-parameter Richards function (Eq. 3). The Richards equation is an extension of the sigmoidal growth curve; specifically, when d coefficient is equal to 1, the sigmoidal and Richards curves are the same. Hence, we analysed the variation of the d coefficient in the presence of different input DNA and PCR efficiencies. Figure 3 shows that the d value is close to 1 at amplification mix percentages ranging from 100% to 90% while at lower amplification mix contents, where PCR efficiency decreases, the d coefficient was significantly higher than 1 regardless of the starting DNA content (Fig. 3; Tab. 2). These data demonstrate that sigmoidal fitting represents a good approximation of real-time PCR kinetic only in the presence of optimal amplification conditions while the Richards curve is more suited when PCR is inhibited. Since the Richards growth equation includes sigmoidal amplification curves, when d = 1, this nonlinear fitting was used in our method.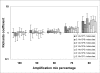
Figure 3
Distribution of Richards coefficients (d) estimated from PCR fluorescence curves using Eq. 3 in nonlinear fitting procedure. Richards coefficient values were determined from 420 independent PCR reactions. The data have been reported in Log10scale, and represented as mean and standard deviation.Table 1
Comparison of five S-shaped models to fit the PCR curve: Sigmoid, Richards, Gompertz, Hill and Chapman. In this table, f is the fluorescence at cycle x; Fmax represents the maximum fluorescence value; Fb is the background reaction fluorescence; b, c and d determine the shape of each curve. For each model the determination coefficient (R2), the adjusted determination coefficient (Adj R2) and the standard error of estimate have been calculated.| Name | Equation | Estimated Parameters | R2 | Adj R2 | Standard Error of Estimate | ||||
| Fmax | b | c | Fb | d | |||||
| Sigmoid | f = Fb+Fmax/(1+exp(-(x-c)/b)) | 45.11 | 1.49 | 22.37 | -0.03 | 1 | 1 | 0.1354 | |
| Richards | f = Fb+(Fmax/(1+exp(-(1/b)*(x-c)))^d) | 45.11 | 1.58 | 21.95 | 0.02 | 1.20 | 1 | 1 | 0.0926 |
| Gompertz | f = Fb+Fmax *exp(-exp(-(x-c)/b)) | 45.19 | 2.15 | 21.45 | 0.29 | 0.9992 | 0.9992 | 0.6006 | |
| Hill | f = Fb+Fmax *x^b/(d^b+x^b) | 45.18 | 14.95 | 0.08 | 22.34 | 1 | 1 | 0.1351 | |
| Chapman | f = Fb+Fmax *(1-exp(-b*x))^d | 45.19 | 0.46 | 0.29 | 20615 | 0.9992 | 0.9992 | 0.6006 | |
Table 2
tstatistic values obtained for all variable combinations. When t < 0 the Richards coefficient is lower than 1, while for t > 0 the Richards coefficient is higher than 1.Significance levels:
* 0.05 <p < 0.01;
** p < 0.01.
| Amplification mix percentage | |||||
|
|
|||||
| Log10input DNA | 100% | 90% | 80% | 70% | 60% |
| 7.5 | 0.28348 | 1.15431 | 2.9303* | 5.43493** | 4.26067** |
| 6.5 | -3.0233* | -0.5329 | 7.8552** | 8.68609** | 7.28178** |
| 5.5 | -2.2195* | 2.70419* | 4.7185** | 8.61406** | 4.60465** |
| 4.5 | 0.97856 | 1.32162 | 2.34* | 16.5192** | 17.5903** |
| 3.5 | 1.00647 | -1.038 | 2.3307* | 13.2572** | 4.65683** |
| 2.5 | -1.731 | -0.5995 | 5.8385** | 6.90378** | 6.13465** |
| 1.5 | 0.14417 | 1.25452 | -0.898 | 1.87978 | 3.69668** |
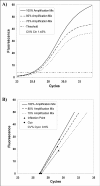
Figure 4
Plot of fluorescence observations versus cycle number obtained from the same starting DNA but in presence of decreasing amounts of amplification mix. This slight PCR inhibition produces curves which are less steep than controls and shifted towards the right. When analysed by the threshold method, these curves showed higher Ct values with a CV% of 1.45% (A). An example of Cy0 procedure has been reported for the same data set (B). In this method, the amplification reactions are described by the tangent crossing the inflection point of fluorescence curves. As shown in this figure, the straight-lines originating from PCRs, characterized by slightly different PCR efficiency and the same starting amounts, tend to cross into a common point near the x-axis leading to small variations in the Cy0 values (CV% = 0.6%).Precision and accuracy of the Ct, Cp and Cy0 methods.
The performance of the Ct, Cp and Cy0 methods was compared in terms of precision and accuracy over a wide range of input DNA concentrations and under different reaction efficiencies obtained by decreasing the amount of amplification mix as reported in Liu and Saint [18, 27]. As shown in Figure 5A, the Ct method is highly rigorous at maximum reaction efficiency regardless of the starting DNA template. However, the absolute value of RE increased almost linearly with the decrease of efficiency regardless of the template concentrations resulting in an underestimation of the unknown of about 50% at the lowest amplification efficiencies. The Cp was more accurate than the Ct method in the presence of different amounts of amplification mix. Indeed, the relative error in the presence of 100% amplification mix tended towards zero as it did using the Ct method. However, when the efficiency declined, the RE increased initially in the same manner at low and high input DNA concentrations, while at 60-70% of the amplification mix, this method markedly underestimated at low concentrations (mean RE60% mix; = -0.58; Fig. 5C). Finally, the Cy0 method was more accurate than the Cp method (mean RE -0.12 versus -0.18, respectively; Fig. 5C, E), which in turn was better than the Ct method (mean RE = -0.31). Notably, at optimal amplification conditions (90-100% of the amplification mix) the Cp and Cy0 methods were equivalent, but at decreasing efficiencies, the Cy0 accuracy was more stable than that of the Cp in the concentration range from 3.14x107 to 3.14x105 molecules. At lower DNA concentrations, from 3.14x104 to 3.14x102 molecules, the RE proportionally increased with the efficiency decline, but this underestimate was less marked than that of the Cp method at the same starting DNA (Fig. 5C, E). Regarding the precision of the three methods, the variation coefficients were determined for each combination of initial template amount and amplification mix percentage. The random error of quantification achieved by the Cp and Cy0 method was similar (mean CV% 21.8% and 22.5%, respectively), while the Ct procedure produced an overall CV% of about 39.7% (Tab. 3). When the CV was analysed in relation to PCR efficiency and input DNA, an area of low variation coefficients for the three methods was found between 3.14x104 and 3.14x107 molecules as starting material (Fig. 5B, D, F). With DNA amounts ranging from 3.14x103 to 3.14x102 molecules, the precision progressively decreased in each analysis procedure. These variations were not efficiency-dependent, but were related to initial DNA quantity as shown by the shapes of level curves reported in figure 5B, D and F, which were perpendicular to the input template amounts.
Figure 5
Comparison of the Ct, Cp and Cy0 methods in terms of precision and accuracy. The accuracy of each method has been reported as Relative Error (RE = expected value ? estimated value) while the precision was evaluated measuring the variation coefficient (CV%). The 3D plots show the variation of relative error in relation to amplification mix percentage and log10 input DNA for the Ct (A), Cp (C) and Cy0 (E) methods. The areas in the level curve graphs represent the CV% values obtained for each amplification mix percentage and Log10 input DNA combination using the Ct (B), Cp (D) and Cy0 (F) methods.Table 3
Comparison of mean Relative Error and mean Variation Coefficient among the Ct, Cp, Cy0 and SCF methods. The reported data were calculated on 420 PCRs except for a) in which the reaction number was 210.| Ct | Cp | Cy0 | SCF | Log10SCF | |
| Mean CV% | 39.70% | 21.80% | 22.52% | 594.74%a | 66.12%a |
| Mean RE | -0.318 | -0.184 | -0.128 | -5.058a | -0.205a |
Experimental system 2: Real-time PCR quantification in the presence of the inhibitor IgG
The real-time amplification plot of 4.05x106 DNA molecules with increasing concentrations of IgG demonstrates the effects of PCR inhibition on amplification efficiency and accumulated fluorescence (Fig. 6A). As inhibitor concentrations increased, the amplification curves showed lower plateau fluorescence levels and a shift towards the right and the bottom of the inflection points, leading to amplification curves that were less steep and not as symmetric as those obtained in absence of the inhibitor agent (Fig. 6A). As shown in figure 6A the amplification curves inhibited by IgG showed a shape very similar to those resulting from the system of amplification mix reduction (system 1; Fig. 4A). Quantitative data analysis of these amplification plots showed that the estimated DNA quantities were systematically underestimated in the presence of IgG concentrations higher than 0.25 µg/ml and 1 µg/ml using Ct and Cp methods, respectively. However, the Cy0 method was able to adjust this bias minimizing the RE at high IgG concentrations (RE = 4.98%; CV = 4.33%; Fig. 6B). Furthermore, in presence of high IgG concentrations, the SCF approach, modified according to Rutledge 2004 [26], was inapplicable because it was impossible to minimize F0 value (Additional file 5).Discussion
None of the current quantitative PCR data treatment methods is in fact fully assumption-free, and their statistical reliability are often poorly characterized. In this study, we evaluated whether known real-time elaboration methods could estimate the amount of DNA in biological samples with precision and accuracy when reaction efficiencies of the unknown are different from those of the standard curve. Our experimental systems consisted in the quantification of samples with the same known starting template amount but the amplification reaction, performed for the real-time PCR assay, had a slightly decreasing efficiency. This is clearly not in agreement with the main assumption of the threshold approach which holds that the amplification efficiency of samples has to be identical to, or not significantly different from, that predicted by the standard curve. However, such an assumption has been reported to be patently invalid for many cases in medical diagnostics. In fact, some, if not all, of the biological samples may contain inhibitors that are not present in the standard nucleic acid samples used to construct the calibration curve, leading to an underestimation of the DNA quantities in the unknown samples [28, 29].In our study, slightly decreasing efficiencies were obtained by two systems: diluting the master enzyme mix or adding IgG, a known inhibitor of PCR. Although, the first system is an "in vitro" simulation of PCR inhibition, it produces amplification curves very similar to those obtained in the presence of a biological inhibitor like IgG. Notably, our experimental setup is not characterized by aberrant amplification reactions. On the contrary, the reactions show a slight mean efficiency decrease which is always the case of biological samples. This PCR inhibition remains undetected when using a threshold approach leading to target underestimation. Moreover, small differences in amplification efficiency produce large quantitative errors and the frequency and magnitude of these errors are virtually impossible to ascertain using a threshold approach. It has been shown that a difference as small as 4% in PCR efficiency could translate into a 400% error in comparative Ct method based quantification [24]. Considering previous works [18, 19] which demonstrated the capability of the SCF method to quantify a sample without prior knowledge of amplification efficiency, our first choice was to process the experimental data by the SCF method. The effectiveness of the SCF approach is based on curve fitting of raw data so that variations unique to each amplification reaction are incorporated into the analysis. Hence, the results reported herein surprisingly demonstrated that the accuracy and precision of the SCF method was markedly impaired when efficiency fell. In fact, when PCR efficiency decreased by about 2.5% (88.8% efficiency value in the presence of 100% of the amplification mix dropped to 84.4% efficiency in the presence of 60% of the mix), we observed, using the SCF method with log-transformation, that the RE and CV went from 15% to 43% and from 61% to 55%, respectively. Furthermore, we found that, when the amplification curve was inhibited, by IgG, the method proposed by Rutledge [26] can not be applied because for each cut-off cycle eliminated from the plateau phase the F0 value progressively decreased without ever reaching a minimum value. These observations are in agreement with two recent studies, which reported that it is possible to obtain absolute quantification from real-time data without a standard curve, but the Ct method remains a gold standard due to the inherent errors of the multiple estimates used in nonlinear regression [19, 20]. These observations are in accordance with Feller ' s conclusions that different S-shaped curves can be effectively fitted with various sigmoid models [30], each providing distinct F0 values. Thus sigmoid fit methods such as the logistic model, used in the SCF approach, are purely descriptive and quantitative results may be unreliable. This led us to develop a new mathematical data treatment method, named Cy0, based on nonlinear regression fitting of real-time fluorescence data. The proposed method ' s main advantages are its use of the Richards equation for obtaining the coordinate of the inflection point and the determination of the quantitative entity Cy0 using the five parameters of reaction curve. Although the logistic growth equation generates a curve that tends towards an exponential form at low fluorescence values, making this curve ideal to model PCR reaction, its maximum slope, or inflection point, is always imposed to be at half the value of the upper asymptote, (Fmax-Fb)/2. This is unsatisfactory because the factors that determine the growth rate are complex and some amplification systems, although characterized by good reaction efficiency, as assessed by standard curve, do not have the center of symmetry in the inflection point. The Richards equation is a more flexible growth function because it has an additional parameter, which is a shape parameter that can make the Richards equation equivalent to the logistic, Gompertz, or monomolecular equations [31]. Variation of the shape parameter allows the point of inflection of the curve to be at any value between the minimum and the upper asymptote; when d = 1 the Eq. 3 becomes the sigmoidal equation. Furthermore, since very small errors of the multiple estimates used in non-linear regression lead to large variations in F0 values, the real-time PCR kinetic parameters were used to define a new quantitative entity, the Cy0. The Cy0 relies on the inflection point position and on the slope of the fluorescence curve at that point, so that its value slightly changes in relation to PCR efficiency. In particular, in a slightly inhibited amplification reaction, the fluorescence curves are shifted towards the right and/or they are less steep; this generates higher Ct values than those found under optimal amplification conditions, underestimating the target amount. In the Cy0 method, the tangents, calculated from different PCR efficiency, tend to intersect at a common point near the x-axis leading to small variations in the Cy0 values (Fig. 4).
The standard curve approach was chosen for the proposed method because currently there no genuine mathematical model for PCR efficiency assessment. The main complication is that actual efficiency amplification is not constant through the PCR run being high in exponential phase and gradually declining towards the plateau phase [33, 35]. However, most current methods of PCR efficiency assessment report "overall" efficiency as a single value [13, 24, 36, 37]. Moreover, recent publications on PCR efficiency assessment have concentrated on the analysis of individual shapes of fluorescence plots in order to estimate a dynamic efficiency value [19, 20, 27, 38]. This proliferation of new methods to assess PCR efficiency demonstrates that, at present, there is not an accepted procedure to evaluate PCR efficiency from a single run, hence some methods can "overestimate" and others "underestimate" the "true" PCR efficiency [8]. In contrast, the standard curve method is based on a simple approximation of data obtained in standard dilutions to unknown samples. In this procedure PCR efficiency assessment is based on the slope of the standard curve. Indeed, the original method (Ct) does not account for PCR efficiencies in individual target samples. The proposed procedure overcomes this limitation by evaluating single amplification variations using Richards curve fitting and subsequently produces a Cy0 value that minimizes the dependence of its value on PCR kinetic. We then compared our method with the Ct method, the actual "gold standard" in real-time PCR quantification and the Cp method which is also used in molecular diagnostics. Both methods are based on standard curve methodology and are the most frequently used in this field. The Ct, Cp and Cy0 methods were evaluated on the same data set using two criteria: precision and accuracy. We defined the accuracy of a model as its ability to provide expected concentrations of the known dilutions under different PCR amplification efficiencies. On the contrary, precision is related to the variability of the results obtained from a given model, and it indicates whether reliable results may be obtained from a small data collection. Our results clearly demonstrated that, under optimal amplification conditions, these three methods were equally precise and accurate. However, when the PCR efficiency decreased, due to amplification mix dilution or IgG presence, the Ct method was markedly impaired and the Cp and Cy0 methods proved to be significantly more accurate than the Ct method. Notably, the Cy0 method showed accuracy levels higher than the Cp method maintaining the same precision. The ability to carry out reliable nuclei acid quantification even in sub-optimal amplification conditions is particularly useful when PCR optimization is not possible, as in the case of high-throughput screening of gene expression or biological samples difficult to cleanse of PCR inhibitors. Furthermore, the Cy0 method is completely objective and assumption-free. Indeed, it does not require the choice of a threshold value and the assumption of similar amplification efficiency between the standard curve and biological samples, necessary in the Ct method. Moreover, there is no need to assume that base pair composition and amplicon size do not impact the fluorescence characteristics of SYBR Green I, required in optical calibration methods like SCF [19]. Our procedure may have future applications in TaqMan assays, where the Taq DNA polymerase digests a probe labelled with a fluorescent reporter and quencher dye and the signal diverges from the product resulting in non-symmetric amplification curves that can be effectively modelled by Richards equation [39]. Further work is needed to extensively verify the accuracy and precision of the Cy0 method in the presence of other known PCR inhibitors like phenol, haemoglobin, fat and tannic acid [17, 22].
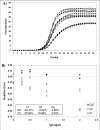
Figure 6
Real-time PCR amplification plots obtained from the same starting DNA in the presence of IgG acting as reaction inhibitor This inhibition system produces curves which are progressively less steep than non-inhibited reactions with increasing IgG concentrations (A). When analysed by the Ct, Cp and Cy0 methods these curves showed a RE% of -25.37%, -9.02% and 4.98% and a CV% of 25.62%, 10.66% and 4.33%%, respectively (B).Conclusions
Real-time PCR analysis is becoming increasingly important in biomedical research because of its accuracy, sensitivity and high efficiency. Although, real-time PCR analysis has gained considerable attention, it is far from being a standard assay. The standard methods are quite stable and straightforward but the accuracy of estimates is strongly impaired if efficiency is not equal in all reactions. Furthermore, the assumption of uniform efficiency has been reported to be invalid in many cases regarding medical diagnostics. In fact, the biological samples may contain inhibitors that could lead to different amplification efficiencies among samples. We propose, in this report, a modified standard curve-based method, called Cy0, that does not require the assumption of uniform reaction efficiency between standards and unknown.List of abbreviations used
Cp: crossing point; Ct: threshold cycle; CV: coefficient of variation; IgG: immunoglobulin G; RE: relative error; SCF: sigmoidal curve fitting.Authors' contributions
MG and DS carried out the design of the study, participated in data analysis, developed the Cy0 method and drafted the manuscript. MBLR participated in data collection and analysis and critically revised the manuscript. LS carried out the real-time PCR. VS participated in the design of the study and critically revised the manuscript. All authors read and approved the final manuscript.Acknowledgements
We thank Dr. Pasquale Tibollo for technical assistance and Dr. Giosué Annibalini for helpful comments on the manuscript.References
1. Higuchi R, Fockler C, Dollinger G, Watson R: Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y) 1993, 11(9):1026-1030.
2. Schmittgen TD: Real-time quantitative PCR. Methods 2001, 25(4):383-385.
3. Bustin SA, Nolan T: Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech 2004, 15(3):155-166.
4. Gingeras TR, Higuchi R, Kricka LJ, Lo YM, Wittwer CT: Fifty years of molecular (DNA/RNA) diagnostics. Clin Chem 2005, 51(3):661-671.
5. Bustin SA, Mueller R: Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin Sci (Lond) 2005, 109(4):365-379.
6. Nolan T, Hands RE, Bustin SA: Quantification of mRNA using real-time RT-PCR. Nature Protocols 2006, 1(3):1559-1582.
7. Marubini E, Verderio P, Raggi CC, Pazzagli M, Orlando C: Statistical diagnostics emerging from external quality control of real-time PCR. The International journal of biological markers 2004, 19(2):141-146.
8. Pfaffl M: Quantification strategies in real time PCR. In A-Z of quantitative PCR Edited by: Bustin SA La Jolla, CA, International University Line 2004.
9. Luu-The V, Paquet N, Calvo E, Cumps J: Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. BioTechniques 2005, 38(2):287-293.
10. Livak KJ: ABI Prism 7700 sequence detection system. User Bulletin 2. PE Applied Biosystems 1997.
11. Rutledge RG, Cote C: Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res 2003, 31(16):e93.
12. Raeymaekers L: A commentary on the practical applications of competitive PCR. Genome Res 1995, 5(1):91-94.
13. Bar T, Stahlberg A, Muszta A, Kubista M: Kinetic Outlier Detection (KOD) in real-time PCR. Nucleic Acids Res 2003, 31(17):e105.
14. Lefevre J, Hankins C, Pourreaux K, Voyer H, Coutlee F: Prevalence of selective inhibition of HPV-16 DNA amplification in
cervicovaginal lavages. Journal of
medical virology 2004, 72(1):132-137.
15. Sunen E, Casas N, Moreno B, Zigorraga C: Comparison of two methods for the detection of hepatitis A virus in clam samples (Tapes spp.) by reverse transcription-nested PCR. International journal of food microbiology 2004, 91(2):147-154.
16. Jiang J, Alderisio KA, Singh A, Xiao L: Development of procedures for direct extraction of Cryptosporidium DNA from water concentrates and for relief of PCR inhibitors. Applied and environmental microbiology 2005, 71(3):1135-1141.
17. Kontanis
EJ, Reed FA: Evaluation of real-time PCR
amplification efficiencies to detect PCR inhibitors. J Forensic Sci 2006, 51(4):795-804.
18. Liu
W, Saint DA: Validation of a
quantitative method for real time PCR kinetics. Biochem Biophys Res Commun 2002, 294(2):347-353.
19. Goll
R, Olsen T, Cui G, Florholmen J: Evaluation
of absolute quantitation by nonlinear regression in probe-based real-time PCR.
BMC Bioinformatics 2006, 7:107.
20. Karlen
Y, McNair A, Perseguers S, Mazza C, Mermod N: Statistical significance of quantitative PCR. BMC Bioinformatics 2007, 8:131.
21. Bustin
SA: Absolute quantification of mRNA
using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 2000, 25(2):169-193.
22. Tichopad
A, Didier A, Pfaffl MW: Inhibition of
real-time RT-PCR quantification due to tissue-specific contaminants. Mol Cell Probes 2004, 18(1):45-50.
23. Nolan
T, Hands RE, Ogunkolade W, Bustin SA: SPUD:
a quantitative PCR assay for the detection of inhibitors in nucleic acid
preparations. Anal Biochem 2006, 351(2):308-310.
24. Ramakers
C, Ruijter JM, Deprez RH, Moorman AF: Assumption-free
analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 2003, 339(1):62-66.
25. Richards
F: A flexible growth function for
empirical use. Journal of
experimental Botany 1959, 10:290-300.
26. Rutledge
RG: Sigmoidal curve-fitting redefines
quantitative real-time PCR with the prospective of developing automated
high-throughput applications. Nucleic
Acids Res 2004, 32(22):e178.
27. Alvarez
MJ, Vila-Ortiz GJ, Salibe MC, Podhajcer OL, Pitossi FJ: Model based analysis of real-time PCR data from DNA binding dye
protocols. BMC Bioinformatics 2007,
8(1):85.
28. Meijerink
J, Mandigers C, van de Locht L, Tonnissen E, Goodsaid F, Raemaekers J: A novel method to compensate for different
amplification efficiencies between patient DNA samples in quantitative
real-time PCR. J Mol Diagn 2001, 3(2):55-61.
29. Stahlberg
A, Aman P, Ridell B, Mostad P, Kubista M: Quantitative
real-time PCR method for detection of B-lymphocyte monoclonality by comparison
of kappa and lambda immunoglobulin light chain expression. Clin Chem 2003, 49(1):51-59.
30. Feller
W: On the logistic law of growth and its
empirical verification in biology. Acta
Bioth Ser A 1940(2):51-66.
31. Birch
C: A new generalized logistic sigmoid
growth equation compared with the Richards growth equation. Annals of Botany 1999, 83:713-723.
32. Yin
X, Goudriaan J, Lantinga EA, Vos J, Spiertz HJ: A flexible sigmoid function of determinate growth. Ann Bot (Lond) 2003, 91(3):361-371.
33. Lalam
N: Estimation of the reaction efficiency
in polymerase chain reaction. J Theor
Biol 2006, 242(4):947-953.
34. Zhao
S, Fernald RD: Comprehensive algorithm
for quantitative real-time polymerase chain reaction. J Comput Biol 2005, 12(8):1047-1064.
35. Gevertz
JL, Dunn SM, Roth CM: Mathematical model
of real-time PCR kinetics. Biotechnol
Bioeng 2005, 92(3):346-355.
36. Peirson
SN, Butler JN, Foster RG: Experimental
validation of novel and conventional approaches to quantitative real-time PCR
data analysis. Nucleic Acids Res 2003,
31(14):e73.
37. Tichopad
A, Dilger M, Schwarz G, Pfaffl MW: Standardized
determination of real-time PCR efficiency from a single reaction set-up. Nucleic Acids Res 2003, 31(20):e122.
38. Liu
W, Saint DA: A new quantitative method
of real time reverse transcription polymerase chain reaction assay based on
simulation of polymerase chain reaction kinetics. Anal Biochem 2002, 302(1):52-59.
39. Livak
KJ, Flood, S.J., Marmaro, J., Giusti, W., Deetz, K.: Oligonucleotides with fluorescent dyes at opposite ends provide a
quenched probe system useful for detecting PCR product and nucleic acid
hybridization. PCR Methods appl 1995,
4:357-362.
Supplementary Material

Windows Excel file containing PCR readouts and non-linear fittings.

Windows Word file containing first and second derivative of Richards equation and the mathematical formulas for obtaining the coordinate of the Cy0 point.
Windows Excel file containing the Ct Cp Cy0 SCF Log10 SCF elaborations.
Windows Excel file containing the results obtained with the SCF approach based on a previous study by Rutledge 2004.
Windows Excel file containing the results obtained with the SCF approach based on a previous study by Rutledge 2004 in presence of IgG.